DR RYAN PATEL BA, PhD
My past and present work has aimed:
1. To understand the neural basis of sensory biomarkers for pain modulation, and to improve the translatability of pre-clinical measures by aligning paradigms in parallel rodent and human studies.
and
2. To identify the neural circuits that mediate the reciprocal interaction between chronic pain and co-morbid mood disorders.
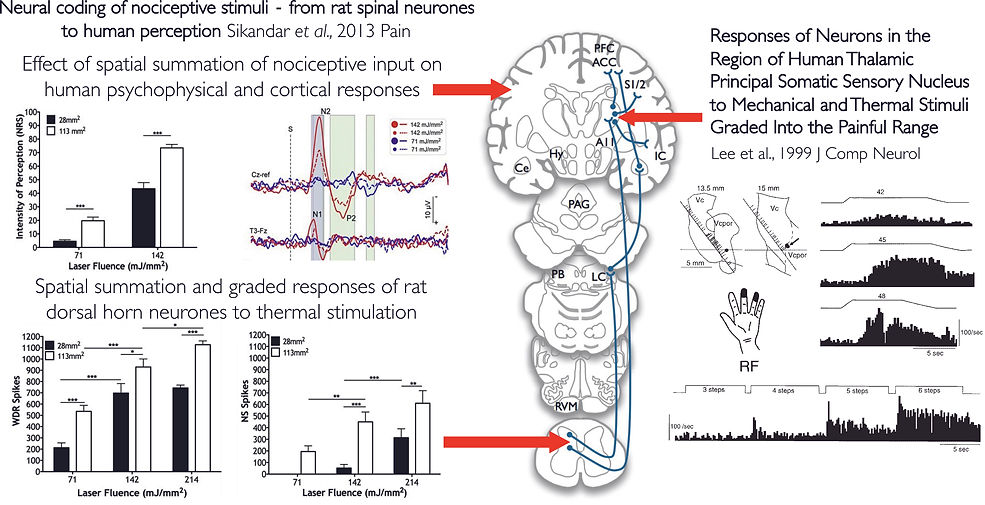
I utilise in vivo electrophysiology to record from deep dorsal horn wide dynamic range neurones within the spinothalamic pathway. Unlike paw withdrawal reflexes which only provide limited insight into response thresholds, neuronal recordings provide a wealth of information about how sensory inputs are processed at the spinal level. These recordings permit the study of: (1) a full stimulus-response relationship and intensity dependent coding, (2) spatial summation, (3) temporal summation (see video below), (4) encoding of stimulus modality, (5) after-firing, (6) local and descending modulation of spinal output, and (7) post-synaptic responses to convergent Aβ, Aδ and C afferent input. These characteristics are conserved across species and single unit recordings have also been obtained from the sensory thalamus of patients undergoing stereotaxic procedures for other conditions. From these we can see that polymodal neurones exist that encode sensory modalities and stimulus intensity, but micro-stimulation at these recording sites also elicit natural sensations in the region of the receptive fields.
WIND-UP
An example of wind-up in the mouse deep dorsal horn. A train of 16 electrical stimuli are delivered at 3x the threshold for activating C-fibres at a frequency of 0.5 Hz.
Note the progressive increase in excitability following repetitive stimulation and the prolonged post-discharges towards the end of the train. Wind-up has been interpreted as the capacity to amplify nociceptive signals and shares many features with central sensitisation.
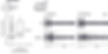
Trendafilova et al., Sodium-Calcium Exchanger-3 Regulates Pain ‘Wind-Up’: From Human Psychophysics to Spinal Mechanisms. Neuron 2022; 110(16):2571-2587.e13 10.1016/j.neuron.2022.05.017
TEMPORAL SUMMATION OF PAIN
Temporal summation of pain describes a perceptual response where repeated low frequency stimulation with a noxious stimulus results in a progressive increase in the perceived pain intensity. Neuronal wind-up in the dorsal horn (above) is thought to represent the spinal neuronal substrate for this perceptual phenomenon. Wind-up is a form of short term neuronal plasticity which generates features of neuronal sensitisation reminiscent of central sensitisation (a longer term form of synaptic plasticity). Temporal summation of pain is frequently interpreted as the capacity to amplify nociceptive inputs and may have value as a sensory biomarker for endogenous pain modulation. Drugs that are capable of inhibiting neuronal wind-up in rodent models may benefit patients with exacerbated temporal summation (e.g. ketamine). Studying temporal summation of pain and wind-up can also aid the identification of novel drug targets, as was the case with NCX3.
LATERAL HABENULA CIRCUITS FOR THE REGULATION OF PAIN AND MOOD
The lateral habenula has long been at the centre of theories linking chronic pain and depression. Habenula hyperactivity has been implicated in numerous psychiatric conditions either indirectly from neuroimaging studies or directly from neuromodulation case studies. The lateral habenula is also proposed as a crucial site of action for the rapid anti-depressant effect of ketamine. As part of its function in encoding aversive states, the lateral habenula is implicated in signalling pain and analgesia. Using viral tracing methods, behaviour and in vivo electrophysiology, I am investigating the role of lateral habenula efferent pathways in regulating mood and the transmission of pain via the descending pain modulatory system in a chronic pain state.
